5月30日,国际权威生物信息期刊《Briefings in Bioinformatics》(IF=13.994)在线发表我院沈其荣院士团队LorMe实验室的最新研究成果《DCiPatho: Deep cross-fusion networks for genome scale identification of pathogens》,研究针对复杂环境微生物组中无法准确识别致病菌污染的难题,利用深度学习构建基于基因组尺度全局性特征的深度交叉融合网络模型DCiPatho,用于人畜和动植物致病菌的精准快速识别,提升环境土壤生物健康的监测能力。
主要内容
致病菌是威胁人类、动植物与环境健康的重要生物性污染因子。如何快速高效检测复杂环境微生物组中的致病菌污染对于公共卫生、动植物检疫和环境质量评估等研究具有重要意义。利用扩增子和宏基因组等测序手段检测致病菌污染主要依赖于数据库的完整性,缺乏对致病菌特征整体性的认识,以及数据库外其他致病菌的识别能力。尽管有研究尝试利用深度学习算法提升致病菌识别的效果,但CNN、Attention和BERT等先进算法以短序列(<3 kb)输入为主,无法直接处理百万bp级别的基因组长序列。当前,主流做法是将长片段DNA作为自然语言切分为若干短序列进行投票预测,忽视了序列之间的语义关系,更缺乏对长片段DNA全局性特征的理解。为解决环境生物健康领域致病菌识别和计算机领域长片段自然语言处理的共性难题,提高基因组尺度致病菌的检测性能,建立了一种基于深度交叉融合网络模型的环境致病菌精准识别系统DCiPatho。
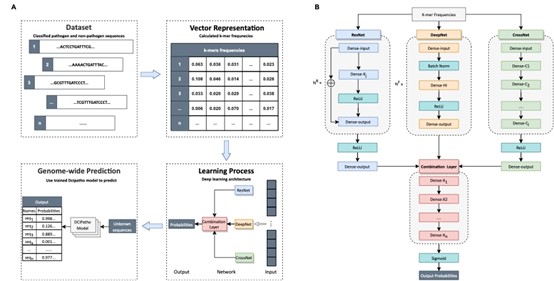
以病原细菌为模式,LorMe实验室联合我校人工智能学院协同攻关,创建了基于交叉融合网络精准识别致病菌的深度学习算法。该研究首先收集了2.2万种致病菌和1.1万种非致病菌,构建了包括7837个属、32,927条细菌的全基因组数据集(BacRefSeq)。据统计,病原细菌的DNA序列长度在1.1 ~ 11.6 Mb之间(中位数3.8.108 bp),非病原细菌的序列长度在4.3 ~ 10.5 Mb之间(中位数3.8.108 bp)。为获得百万bp级别长序列特征,将全基因组3 ~ 7的k-mer频率词向量特征进行全组合,开发了深度交叉融合网络模型算法DCiPatho。该算法将交叉网络、残差网络和深度神经网络进行深度交叉融合,实现高阶融合特征的自动学习,大幅度降低了计算成本。虚拟实验结果表明,DCiPatho的性能超越了Bi-LSTM、CNN、Transformer、DeePaC和BERTax等9种公认的高性能深度学习算法,识别精度高达95.14%。该研究为环境土壤生物健康评估与人畜安全检测提供了新方法、新技术与新途径。
该研究得到国家自然科学基金重大项目和中央高校基本业务费等项目的资助。我院韦中教授与人工智能学院薛卫副教授为共同通讯作者,开云网页版登录入口江高飞副教授为第一作者,人工智能学院研究生张家璇、陈行健(现香港城市大学博士生),开云网页版登录入口博士生张耀中、杨欣润和汪宁祺以及研究生李婷婷参与了该研究。沈其荣院士、赵方杰教授和徐阳春教授共同指导了该项研究。
据悉,我校人工智能学院薛卫组与沈其荣院士团队LorMe实验室合作开展了大量研究工作,在利用人工智能进行基因和蛋白序列编码与功能预测、堆肥腐熟预测、梨树病害检测和根系构型分析等方面取得了系列进展和突破,相关成果在Brief Bioinform、Bioresour Technol、Biomed Res Int等国际权威杂志发表研究论文25篇,获授权/受理国家发明专利8件,软著19套。
全文链接:https://doi.org/10.1093/bib/bbad194
 微信公众号
微信公众号